Scalar field analysis
Once the electronic structure has been determined, a variety of scalar fields derived from the electron density can be analyzed and visualized. These fields provide insight into the spatial distribution of electronic properties and form the basis for interpreting exchange, correlation, and density-related quantities.
Electron density and its gradient
The pydft.DFT class exposes its internal matrices and a number of
useful functions which we can readily use to interpret its operation. Let us
start by generating a plot of the electron density using the method
pydft.DFT.get_density_at_points().
1import numpy as np
2from pydft import MoleculeBuilder, DFT
3import matplotlib.pyplot as plt
4from mpl_toolkits.axes_grid1 import make_axes_locatable
5
6# perform DFT calculation on the CO molecule
7co = MoleculeBuilder().from_name("CO")
8dft = DFT(co, basis='sto3g')
9res = dft.scf(1e-6, verbose=False)
10
11# generate grid of points and calculate the electron density for these points
12sz = 4 # size of the domain
13npts = 100 # number of sampling points per cartesian direction
14
15# produce meshgrid for the xz-plane
16x = np.linspace(-sz/2,sz/2,npts)
17zz, xx = np.meshgrid(x, x, indexing='ij')
18gridpoints = np.zeros((zz.shape[0], xx.shape[1], 3))
19gridpoints[:,:,0] = xx
20gridpoints[:,:,2] = zz
21gridpoints[:,:,1] = np.zeros_like(xx) # set y-values to 0
22gridpoints = gridpoints.reshape((-1,3))
23
24# calculate (logarithmic) scalar field and convert if back to an 2D array
25density = dft.get_density_at_points(gridpoints)
26density = np.log10(density.reshape((npts, npts)))
27
28# build contour plot
29fig, ax = plt.subplots(1,1, dpi=144, figsize=(4,4))
30im = ax.contourf(x, x, density, levels=np.linspace(-3,3,13, endpoint=True))
31ax.contour(x, x, density, colors='black', levels=np.linspace(-3,3,13, endpoint=True))
32ax.set_aspect('equal', 'box')
33ax.set_xlabel('x-coordinate [a.u.]')
34ax.set_ylabel('z-coordinate [a.u.]')
35divider = make_axes_locatable(ax)
36cax = divider.append_axes('right', size='5%', pad=0.05)
37fig.colorbar(im, cax=cax, orientation='vertical')
38ax.set_title('Electron density')
39plt.show()
Running the above script yields the electron density.
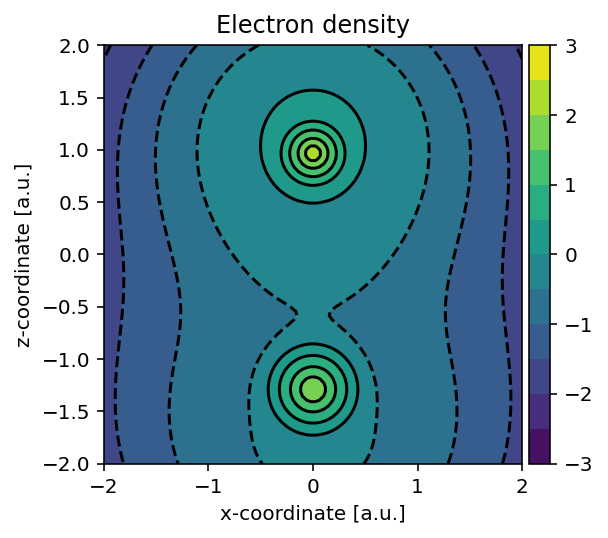
Logarithmic electron density of CO sampled in the molecular plane.
Using largely the same code, we can also readily build the electron density
gradient magnitude using pydft.DFT.get_gradient_at_points().
1import numpy as np
2from pydft import MoleculeBuilder, DFT
3import matplotlib.pyplot as plt
4from mpl_toolkits.axes_grid1 import make_axes_locatable
5
6# perform DFT calculation on the CO molecule
7co = MoleculeBuilder().from_name("CO")
8dft = DFT(co, basis='sto3g')
9res = dft.scf(1e-6, verbose=False)
10
11# generate grid of points and calculate the electron density for these points
12sz = 4 # size of the domain
13npts = 100 # number of sampling points per cartesian direction
14
15# produce meshgrid for the xz-plane
16x = np.linspace(-sz/2,sz/2,npts)
17zz, xx = np.meshgrid(x, x, indexing='ij')
18gridpoints = np.zeros((zz.shape[0], xx.shape[1], 3))
19gridpoints[:,:,0] = xx
20gridpoints[:,:,2] = zz
21gridpoints[:,:,1] = np.zeros_like(xx) # set y-values to 0
22gridpoints = gridpoints.reshape((-1,3))
23
24# calculate (logarithmic) scalar field and convert if back to an 2D array
25gradient = np.linalg.norm(dft.get_gradient_at_points(gridpoints), axis=1)
26gradient = np.log10(gradient.reshape((npts, npts)))
27
28# build contour plot
29fig, ax = plt.subplots(1,1, dpi=144, figsize=(4,4))
30im = ax.contourf(x, x, gradient, levels=np.linspace(-2,4,7, endpoint=True))
31ax.contour(x, x, gradient, colors='black', levels=np.linspace(-2,4,7, endpoint=True))
32ax.set_aspect('equal', 'box')
33ax.set_xlabel('x-coordinate [a.u.]')
34ax.set_ylabel('z-coordinate [a.u.]')
35divider = make_axes_locatable(ax)
36cax = divider.append_axes('right', size='5%', pad=0.05)
37fig.colorbar(im, cax=cax, orientation='vertical')
38ax.set_title('Electron density gradient magnitude')
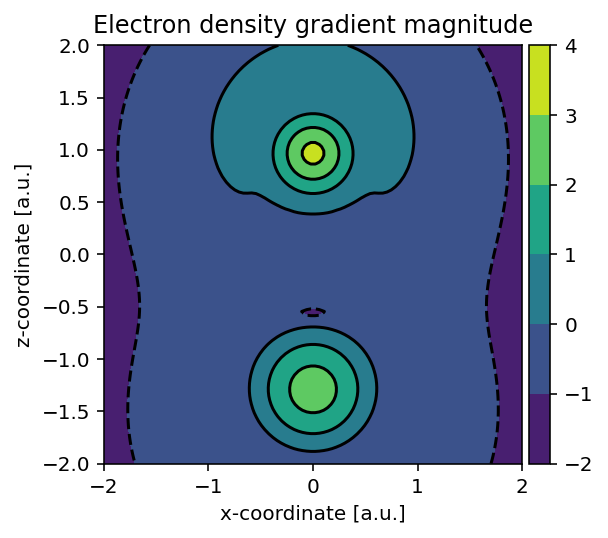
Magnitude of the electron-density gradient in the same plane.
Exchange potential
To construct the exchange potential at arbitrary grid points, we can use the
script as shown below which uses the pydft.MolecularGrid.get_exchange_potential_at_points()
method.
1from pydft import DFT
2from pyqint import MoleculeBuilder
3import numpy as np
4import matplotlib.pyplot as plt
5from mpl_toolkits.axes_grid1 import make_axes_locatable
6
7# perform DFT calculation on the CO molecule
8co = MoleculeBuilder().from_name("CO")
9dft = DFT(co, basis='sto3g')
10res = dft.scf(1e-6, verbose=False)
11
12# grab molecular orbital energies and coefficients
13orbc = res['orbc']
14orbe = res['orbe']
15
16# generate grid of points and calculate the electron density for these points
17sz = 4 # size of the domain
18npts = 100 # number of sampling points per cartesian direction
19
20# produce meshgrid for the xz-plane
21x = np.linspace(-sz/2,sz/2,npts)
22zz, xx = np.meshgrid(x, x, indexing='ij')
23gridpoints = np.zeros((zz.shape[0], xx.shape[1], 3))
24gridpoints[:,:,0] = xx
25gridpoints[:,:,2] = zz
26gridpoints[:,:,1] = np.zeros_like(xx) # set y-values to 0
27gridpoints = gridpoints.reshape((-1,3))
28
29# grab a copy of the MolecularGrid object
30molgrid = dft.get_molgrid_copy()
31
32# construct exchange potential
33field = molgrid.get_exchange_potential_at_points(gridpoints, res['density'])
34field = field.reshape((npts, npts))
35
36# plot field
37fig, ax = plt.subplots(1, 1, dpi=144, figsize=(4,4))
38levels = np.linspace(-6,6,25, endpoint=True)
39im = ax.contourf(x, x, field, levels=levels, cmap='PiYG')
40ax.contour(x, x, field, levels=levels, colors='black', linewidths=0.5)
41ax.set_aspect('equal', 'box')
42ax.set_xlabel('x-coordinate [a.u.]')
43ax.set_ylabel('z-coordinate [a.u.]')
44divider = make_axes_locatable(ax)
45cax = divider.append_axes('right', size='5%', pad=0.05)
46fig.colorbar(im, cax=cax, orientation='vertical')
47ax.set_title('Exchange potential')
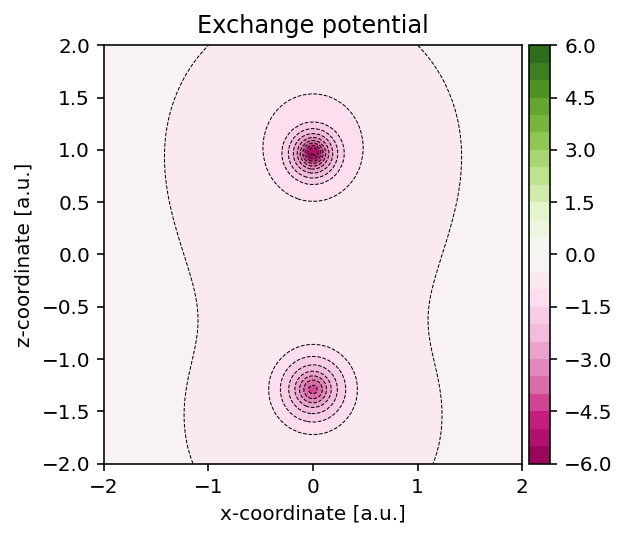
Exchange potential evaluated on an arbitrary two-dimensional grid.
Correlation potential
In a similar fashion as for the exchange potential, we can use the method
pydft.MolecularGrid.get_correlation_potential_at_points() to obtain the
correlation potential field.
1from pydft import DFT
2from pyqint import MoleculeBuilder
3import numpy as np
4import matplotlib.pyplot as plt
5from mpl_toolkits.axes_grid1 import make_axes_locatable
6
7# perform DFT calculation on the CO molecule
8co = MoleculeBuilder().from_name("CO")
9dft = DFT(co, basis='sto3g')
10res = dft.scf(1e-6, verbose=False)
11
12# grab molecular orbital energies and coefficients
13orbc = res['orbc']
14orbe = res['orbe']
15
16# generate grid of points and calculate the electron density for these points
17sz = 4 # size of the domain
18npts = 100 # number of sampling points per cartesian direction
19
20# produce meshgrid for the xz-plane
21x = np.linspace(-sz/2,sz/2,npts)
22zz, xx = np.meshgrid(x, x, indexing='ij')
23gridpoints = np.zeros((zz.shape[0], xx.shape[1], 3))
24gridpoints[:,:,0] = xx
25gridpoints[:,:,2] = zz
26gridpoints[:,:,1] = np.zeros_like(xx) # set y-values to 0
27gridpoints = gridpoints.reshape((-1,3))
28
29# grab a copy of the MolecularGrid object
30molgrid = dft.get_molgrid_copy()
31
32# construct correlation potential
33field = molgrid.get_correlation_potential_at_points(gridpoints, res['density'])
34field = field.reshape((npts, npts))
35
36# plot field
37fig, ax = plt.subplots(1, 1, dpi=144, figsize=(4,4))
38levels = np.linspace(-0.2,0.2,101, endpoint=True)
39im = ax.contourf(x, x, field, levels=levels, cmap='PiYG')
40ax.contour(x, x, field, levels=levels, colors='black', linewidths=0.5)
41ax.set_aspect('equal', 'box')
42ax.set_xlabel('x-coordinate [a.u.]')
43ax.set_ylabel('z-coordinate [a.u.]')
44divider = make_axes_locatable(ax)
45cax = divider.append_axes('right', size='5%', pad=0.05)
46fig.colorbar(im, cax=cax, orientation='vertical')
47ax.set_title('Correlation potential')
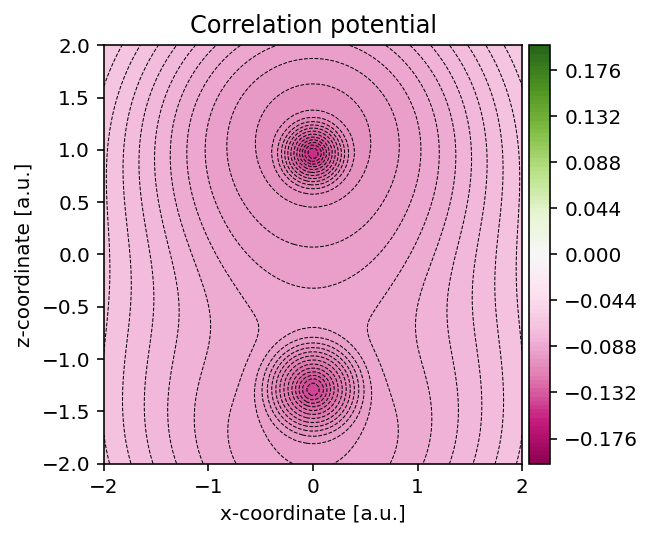
Correlation potential evaluated on the same grid.