Electronic Structure Calculations
To start, we perform a high-level calculation of the electronic structure
of the carbon-monoxide molecule using the default SVWN5 exchange-correlation
functional [Slater, 1951, Vosko et al., 1980].
To perform this calculation, we first have to construct a
pydft.DFT object which requires a molecule as its input. Next, we use
the pydft.DFT.scf() routine to start the self-consistent field calculation.
1from pydft import MoleculeBuilder, DFT
2
3# perform DFT calculation on the CO molecule
4co = MoleculeBuilder().from_name("CO")
5dft = DFT(co, basis='sto3g')
6res = dft.scf(1e-6, verbose=False)
7
8# print total energy
9print("Total electronic energy: %12.6f Ht" % res['energy'])
Performing this calculation shows that the total electronic energy for this system corresponds to:
Total electronic energy: -111.130470 Ht
Result dictionary
The result of an SCF calculation is captured in a Python dictionary object. This dictionary contains all quantities required to analyze, post-process, and validate the electronic structure calculation. The data layout is shared between PyDFT and PyQInt to ensure a consistent interface across methods.
The most commonly used key is energy, which stores the final converged
total energy. Other entries expose the objects that appear in the SCF equations:
the density matrix \(\mathbf{P}\), the one-electron matrices
\(\mathbf{S}\), \(\mathbf{T}\), and \(\mathbf{V}\), the Hartree
matrix \(\mathbf{J}\), and the exchange-correlation matrix. This is why
examples usually store the SCF result as res and then access quantities
such as res['energy'] or res['density'].
Key |
Description |
|---|---|
|
Final converged total electronic energy (Hartree). |
|
Total electronic energy at each SCF iteration. |
|
Electronic kinetic energy, \(\mathrm{Tr}(\mathbf{T}\mathbf{P})\). |
|
Electron-nuclear attraction energy, \(\mathrm{Tr}(\mathbf{V}\mathbf{P})\). |
|
Electron-electron Coulomb (Hartree) energy, \(\mathrm{Tr}(\mathbf{J}\mathbf{P})\). |
|
Nuclear-nuclear repulsion energy. |
|
Exchange energy (Hartree-Fock or DFT). |
|
Correlation energy (DFT only). |
|
Total exchange-correlation energy. |
|
Molecular orbital eigenvalues (orbital energies). |
|
Molecular orbital coefficient matrix (AO basis). |
|
One-particle density matrix \(\mathbf{P}\). |
|
Fock matrix \(\mathbf{F}\). |
|
Hartree (Coulomb) matrix \(\mathbf{J}\). |
|
Exchange-correlation matrix (DFT only, |
|
Overlap matrix \(\mathbf{S}\). |
|
Kinetic energy matrix \(\mathbf{T}\). |
|
Nuclear attraction matrix \(\mathbf{V}\). |
|
Core Hamiltonian matrix \(\mathbf{H}_\mathrm{core} = \mathbf{T} + \mathbf{V}\). |
|
Molecular object defining atoms, geometry, and charge. |
|
Nuclear positions and charges. |
|
List of contracted Gaussian basis functions. |
|
Total number of electrons. |
|
Dictionary containing timing information for construction and SCF iterations. |
Energy decomposition
The molecular matrices can be used to perform a so-called energy decomposition, i.e., decompose the total electronic energy into the kinetic, nuclear attraction, electron-electron repulsion and exchange-correlation energy.
1import numpy as np
2from pydft import MoleculeBuilder, DFT
3
4co = MoleculeBuilder().from_name("CO")
5dft = DFT(co, basis='sto3g')
6res = dft.scf(1e-4, verbose=False)
7print("Total electronic energy: %12.6f Ht" % res['energy'])
8print()
9
10# retrieve molecular matrices
11P = res['density']
12T = res['kinetic']
13V = res['nuclear']
14J = res['hartree']
15
16# calculate energy terms
17Et = np.einsum('ji,ij', P, T)
18Ev = np.einsum('ji,ij', P, V)
19Ej = 0.5 * np.einsum('ji,ij', P, J)
20Ex = res['ex']
21Ec = res['ec']
22Exc = res['exc']
23Enuc = res['enucrep']
24
25print('Kinetic energy: %12.6f Ht' % Et)
26print('Nuclear attraction: %12.6f Ht' % Ev)
27print('Electron-electron repulsion: %12.6f Ht' % Ej)
28print('Exchange energy: %12.6f Ht' % (Ex))
29print('Correlation energy: %12.6f Ht' % (Ec))
30print('Exchange-correlation energy: %12.6f Ht' % (Exc))
31print('Nucleus-nucleus repulsion: %12.6f Ht' % (Enuc))
32print()
33print('Sum: %12.6f Ht' % (Et + Ev + Ej + Exc + Enuc))
The above script yields the following output:
Total electronic energy: -111.130470 Ht
Kinetic energy: 110.217226 Ht
Nuclear attraction: -304.930911 Ht
Electron-electron repulsion: 75.612987 Ht
Exchange energy: -12.055233 Ht
Correlation energy: -1.232632 Ht
Exchange-correlation energy: -13.287865 Ht
Nucleus-nucleus repulsion: 21.258092 Ht
Sum: -111.130470 Ht
Self-consistent field matrices
To visualize the overlap matrix \(\mathbf{S}\) and the Fock matrix \(\mathbf{F}\), we can use the script as found below.
1import numpy as np
2from pydft import MoleculeBuilder, DFT
3import matplotlib.pyplot as plt
4
5def main():
6 # perform DFT calculation on the CO molecule
7 co = MoleculeBuilder().from_name("CO")
8 dft = DFT(co, basis='sto3g')
9 res = dft.scf(1e-6, verbose=False)
10
11 # build list of basis functions
12 labels = []
13 for a in co.get_atoms():
14 for o in ['1s', '2s', '2px', '2py', '2pz']:
15 labels.append('%s - %s' % (a[0],o))
16
17 fig, ax = plt.subplots(1, 2, dpi=144, figsize=(8,4))
18 plot_matrix(ax[0], res['overlap'], xlabels=labels, ylabels=labels, title='Overlap matrix')
19 plot_matrix(ax[1], res['fock'], xlabels=labels, ylabels=labels, title='Hamiltonian matrix')
20
21def plot_matrix(ax, mat, xlabels=None, ylabels=None, title = None, xlabelrot=90):
22 """
23 Produce plot of matrix
24 """
25 ax.imshow(mat, vmin=-1, vmax=1, cmap='PiYG')
26 for i in range(mat.shape[0]):
27 for j in range(mat.shape[1]):
28 ax.text(i, j, '%.2f' % mat[j,i], ha='center', va='center',
29 fontsize=7)
30 ax.set_xticks([])
31 ax.set_yticks([])
32 ax.hlines(np.arange(1, mat.shape[0])-0.5, -0.5, mat.shape[0] - 0.5,
33 color='black', linestyle='--', linewidth=1)
34 ax.vlines(np.arange(1, mat.shape[0])-0.5, -0.5, mat.shape[0] - 0.5,
35 color='black', linestyle='--', linewidth=1)
36
37 ax.set_xticks(np.arange(0, mat.shape[0]))
38 if xlabels is not None:
39 ax.set_xticklabels(xlabels, rotation=xlabelrot)
40 ax.set_yticks(np.arange(0, mat.shape[0]))
41
42 if ylabels is not None:
43 ax.set_yticklabels(ylabels, rotation=0)
44 ax.tick_params(axis='both', which='major', labelsize=7)
45
46 if title is not None:
47 ax.set_title(title)
48
49if __name__ == '__main__':
50 main()
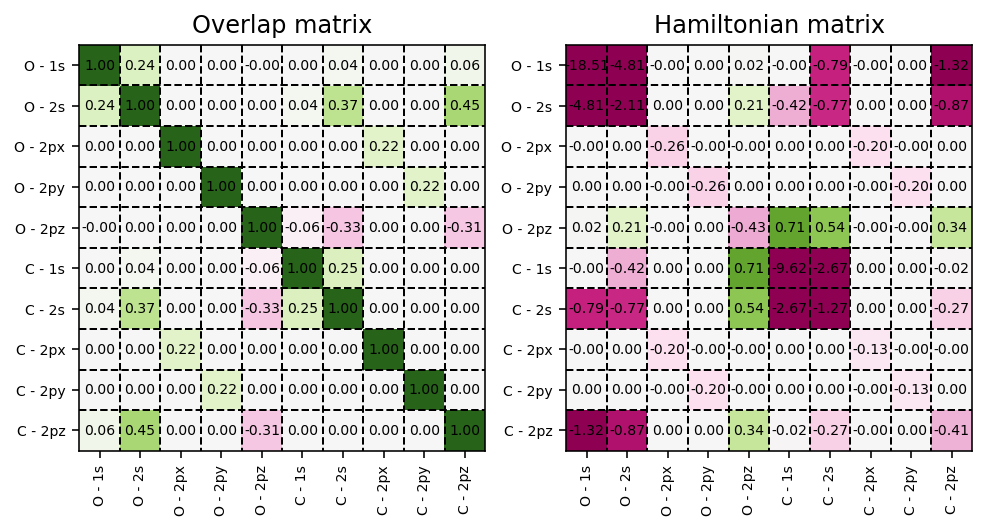
Overlap and Hamiltonian matrices exposed by the SCF result dictionary.
Showing the electronic steps
To get verbose output, i.e. information per electronic step, configure
logging and specify verbose = True.
Note
PyDFT uses Python’s standard logging module for these progress messages
instead of printing directly. This means that verbose = True tells
PyDFT to create informational log messages, while
logging.basicConfig(...) tells Python where and how to show them.
Without the logging configuration, the calculation still runs normally,
but the progress messages remain hidden. This makes it possible to use PyDFT
quietly in scripts, tests, and notebooks, or to send messages to the terminal
when interactive feedback is useful.
1import logging
2
3from pydft import MoleculeBuilder, DFT
4
5logging.basicConfig(level=logging.INFO, format="%(message)s")
6
7# perform DFT calculation on the CO molecule
8co = MoleculeBuilder().from_name("CO")
9dft = DFT(co, basis='sto3g')
10res = dft.scf(1e-5, verbose=True)
Executing the script above yields output like the following. The timings depend on the machine and runtime environment:
001 | E = -179.237419 | dE = 0.0000e+00 | 0.0010 s
002 | E = -106.662588 | dE = 0.0000e+00 | 1.2765 s
003 | E = -117.641796 | dE = 7.2575e+01 | 0.0172 s
004 | E = -107.190988 | dE = 1.0979e+01 | 0.0201 s
005 | E = -117.376118 | dE = 1.0451e+01 | 0.0203 s
006 | E = -117.085797 | dE = 1.0185e+01 | 0.0160 s
007 | E = -108.013652 | dE = 2.9032e-01 | 0.0149 s
008 | E = -107.421366 | dE = 9.0721e+00 | 0.0143 s
009 | E = -110.457951 | dE = 5.9229e-01 | 0.0151 s
010 | E = -110.419024 | dE = 3.0366e+00 | 0.0176 s
011 | E = -109.548780 | dE = 3.8927e-02 | 0.0159 s
012 | E = -111.008079 | dE = 8.7024e-01 | 0.0159 s
013 | E = -111.118875 | dE = 1.4593e+00 | 0.0163 s
014 | E = -111.130700 | dE = 1.1080e-01 | 0.0162 s
015 | E = -111.130454 | dE = 1.1825e-02 | 0.0210 s
016 | E = -111.130470 | dE = 2.4610e-04 | 0.0258 s
017 | E = -111.130470 | dE = 1.6288e-05 | 0.0188 s
018 | E = -111.130470 | dE = 9.2335e-08 | 0.0158 s
Stopping SCF cycle, convergence reached.
Each line corresponds to one update of the density matrix. Internally, PyDFT
uses the current density matrix to build the electron density on the molecular
grid, constructs the Hartree and exchange-correlation matrices, forms the
Kohn-Sham/Fock matrix, diagonalizes it, and then builds the next density matrix
from the occupied orbitals. The energy difference dE is the convergence
measure used by pydft.DFT.scf().
Different exchange-correlation functionals
To use a different exchange-correlation functional, pass the functional
argument when constructing the pydft.DFT object:
1from pydft import MoleculeBuilder, DFT
2
3# perform DFT calculation on the CO molecule
4co = MoleculeBuilder().from_name("CO")
5
6# use SVWN XC functional
7dft = DFT(co, basis='sto3g', functional='svwn5')
8print('SVWN: ', dft.scf(1e-5)['energy'], 'Ht')
9
10# use PBE XC functional
11dft = DFT(co, basis='sto3g', functional='pbe')
12print('PBE: ', dft.scf(1e-5)['energy'], 'Ht')
which yields the following total electronic energies for the SVWN5 and
PBE [Perdew et al., 1996] exchange-correlation functions:
SVWN: -111.13047044798054 Ht
PBE: -111.64036457334683 Ht
Tuning the numerical accuracy
The numerical accuracy of an electronic structure calculation in
PyDFT can be controlled through the specification of the radial and
angular integration grids. These grids are defined per atomic species and can be
adjusted by the user when constructing the pydft.DFT object.
The radial grid is controlled via the number of radial shells
(nshells), while the angular resolution is controlled via the number of
angular points (nangpts). Both parameters are provided as mappings from
element symbols to integer values. Increasing either parameter generally
improves the accuracy of the numerical integration at the cost of increased
computational effort.
The angular grid in PyDFT is based on Lebedev quadrature. As a
consequence, the number of angular points must correspond to one of the
predefined Lebedev grid sizes [Lebedev, 1976]. Arbitrary values for
nangpts are not permitted; only the fixed values listed below are valid.
The supported numbers of angular points are:
6, 14, 26, 38, 50, 74, 86, 110, 146, 170, 194, 230,
266, 302, 350, 434, 590, 770, 974, 1202, 1454, 1730,
2030, 2354, 2702, 3074, 3470, 3890, 4334, 4802,
5294, 5810
By default, PyDFT uses 20, 25, and 30 radial shells for first-,
second-, and third-row atoms, respectively. For the angular grid, 110 angular
points are used for all atoms, except for hydrogen, for which 50 angular points
are used. The maximum angular momentum quantum number lmax is determined
automatically from the chosen number of angular points; for the default values,
this corresponds to lmax = 8 for 110 angular points and
lmax = 5 for 50 angular points.
Users may override these defaults by explicitly specifying nshells and
nangpts when constructing the pydft.DFT object. For example, a
calculation with a moderately accurate grid can be set up as
1from pydft import MoleculeBuilder, DFT
2
3mol = MoleculeBuilder().from_name("CO")
4dft = DFT(mol, basis='sto3g',
5 nshells={'C': 32, 'O': 32},
6 nangpts={'C': 230, 'O': 230}
7)
8res = dft.scf(1e-6, verbose=False)
9
10print("Total electronic energy: %12.6f Ht" % res['energy'])
which shows the following output:
Total electronic energy: -111.142418 Ht
If higher accuracy is required, the number of radial shells can be increased, for example to 64, and the number of angular points can be increased by selecting a larger Lebedev grid from the list above. In practice, we observe that increasing the number of angular points beyond a certain threshold does not lead to significant further improvements in accuracy, whereas increasing the number of radial shells continues to systematically reduce the error.
By adjusting nshells and selecting an appropriate Lebedev grid via
nangpts, users can balance computational cost and numerical accuracy
according to the requirements of their specific application.