Becke grid analysis
All numerical integrations are performed by means of Gauss-Chebychev and Lebedev quadrature using the Becke grid [Becke, 1988]. The radial Gauss-Chebychev points describe the distance from an atomic center, while the Lebedev points [Lebedev, 1976] describe directions on the unit sphere. Their tensor product gives an atom-centered grid.
For molecules, these atom-centered grids overlap. Becke weights turn the overlap into a smooth partition of unity: each grid point receives one weight per atom, and the weights at that point sum to one. Integrals over the whole molecule can therefore be evaluated as a weighted sum of atomic-grid contributions.
Atomic fuzzy cells
It is possible to produce a contour plot of
the fuzzy cells (molecular weights) on a plane using the
pydft.MolecularGrid.calculate_weights_at_points() method. An example
is provided below.
1from pydft import MolecularGrid
2from pyqint import MoleculeBuilder
3import numpy as np
4import matplotlib.pyplot as plt
5
6# construct molecule
7mol = MoleculeBuilder().from_name('benzene')
8cgfs, atoms = mol.build_basis('sto3g')
9
10# construct molecular grid
11molgrid = MolecularGrid([a for a in mol], cgfs,
12 nshells = {'C' : 32, 'H' : 32},
13 nangpts = {'C' : 194, 'H' : 194}
14 )
15
16# produce grid of sampling points to calculate the atomic
17# weight coefficients for
18N = 150
19sz = 8
20x = np.linspace(-sz,sz,N)
21xv,yv = np.meshgrid(x,x)
22points = np.array([[x,y,0] for x,y in zip(xv.flatten(),yv.flatten())])
23
24# calculate the atomic weights
25mweights = molgrid.calculate_weights_at_points(points, k=3)
26
27# plot the atomic weights
28plt.figure(dpi=144)
29plt.imshow(np.max(mweights,axis=0).reshape((N,N)),
30 extent=(-sz,sz,-sz,sz), interpolation='bicubic')
31plt.xlabel('x [a.u.]')
32plt.ylabel('y [a.u.]')
33plt.colorbar()
34plt.grid(linestyle='--', color='black', alpha=0.5)
35
36# add the atoms to the plot
37r = np.zeros((len(atoms), 3))
38for i,at in enumerate(atoms):
39 r[i] = at[0]
40plt.scatter(r[0:6,0], r[0:6,1], s=50.0, color='grey', edgecolor='black')
41plt.scatter(r[6:12,0], r[6:12,1], s=50.0, color='white', edgecolor='black')
42
43plt.tight_layout()
In the script above, we color every point by the maximum value for each of the atomic weights. When this maximum value is one, this implies that the grid point belongs to a single atom. When multiple atoms ‘share’ a grid point, the maximum value among the atomic weights will be lower than one.
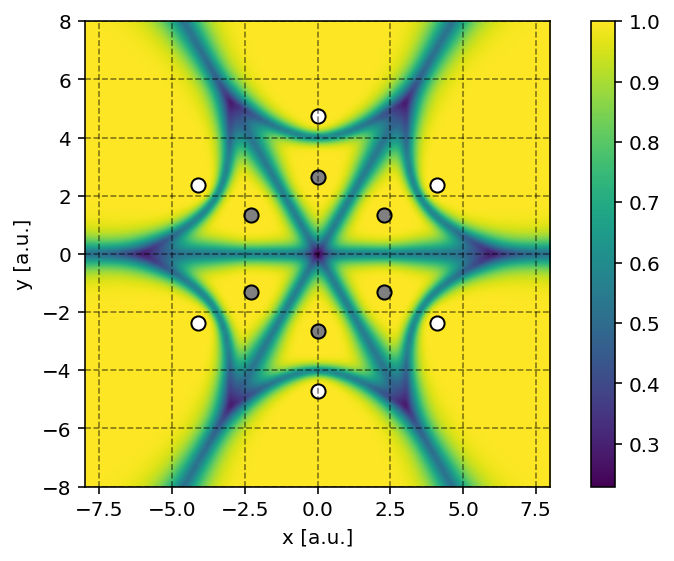
Becke fuzzy-cell weights for benzene projected onto the molecular plane.
Note
Producing such a contour plot is only meaningful for planar molecules such as benzene. For more complex molecules such as methane, it is rather difficult to make sense of the fuzzy cells upon projection on a plane.
Grid points
To obtain the set of grid points on which the numerical integration (quadrature),
is performed, we can invoke the pydft.MolecularGrid.get_grid_coordinates()
method.
1from pydft import MolecularGrid
2from pyqint import MoleculeBuilder
3import matplotlib.pyplot as plt
4
5# construct molecule
6mol = MoleculeBuilder().from_name('co')
7cgfs, atoms = mol.build_basis('sto3g')
8
9# construct molecular grid
10molgrid = MolecularGrid([a for a in mol], cgfs,
11 nshells = {'C' : 32, 'O' : 32},
12 nangpts = {'C' : 50, 'O' : 50},
13 lmax = {'C' : 5, 'O' : 5}
14 )
15molgrid.initialize()
16
17# obtain the grid points
18gridpoints = molgrid.get_grid_coordinates()
19
20# plot the atomic weights
21colors = '#DD0000', '#222222'
22fig = plt.figure(dpi=300, figsize=(8,6))
23ax = fig.add_subplot(projection='3d')
24for i in range(0, len(gridpoints)):
25 ax.scatter(gridpoints[i][:,0], gridpoints[i][:,1], gridpoints[i][:,2],
26 s = 1.5, alpha=0.5, color=colors[i])
27
28# set axes
29ax.set_xlim(-5,5)
30ax.set_ylim(-5,5)
31ax.set_zlim(-5,5)
32ax.set_xlabel('x [a.u.]')
33ax.set_ylabel('y [a.u.]')
34ax.set_zlabel('z [a.u.]')
35ax.set_box_aspect(aspect=None, zoom=0.8)
36
37plt.show()
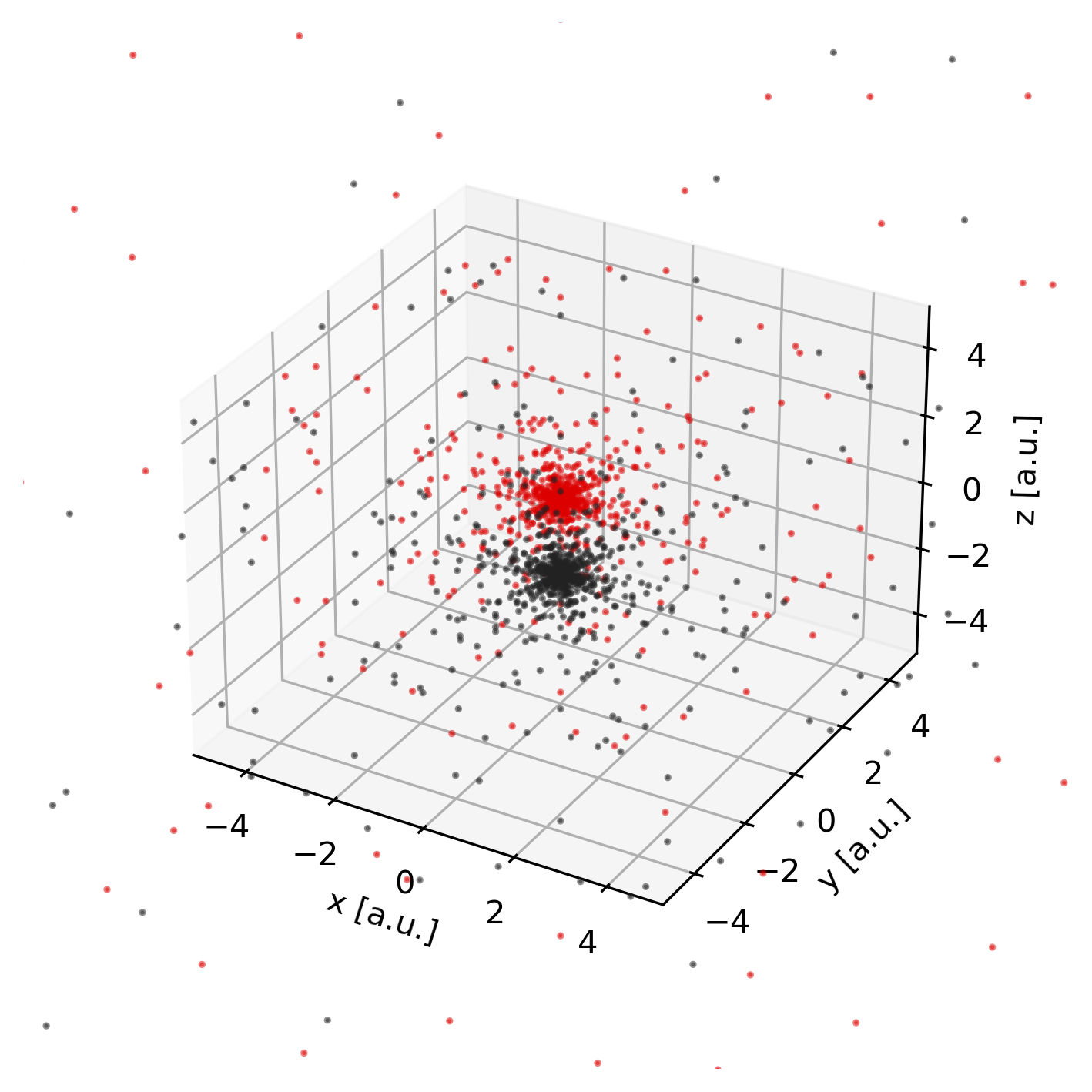
Atom-centered quadrature points used to perform the molecular integration.