Orbital Visualization
Within the Kohn-Sham approximation, the set of molecular orbitals that minimizes the total electronic energy are found via the diagonalization of the Fock matrix.
Coefficient matrix
These Kohn-Sham orbitals are encoded in the coefficient matrix which is extracted via the script below.
1import numpy as np
2from pydft import MoleculeBuilder, DFT
3import matplotlib.pyplot as plt
4
5def main():
6 # perform DFT calculation on the CO molecule
7 co = MoleculeBuilder().from_name("CO")
8 dft = DFT(co, basis='sto3g')
9 res = dft.scf(1e-6, verbose=False)
10
11 # build list of basis functions
12 labels = []
13 for a in co.get_atoms():
14 for o in ['1s', '2s', '2px', '2py', '2pz']:
15 labels.append('%s - %s' % (a[0],o))
16
17 fig, ax = plt.subplots(1, 1, dpi=144, figsize=(4,4))
18 plot_matrix(ax, res['orbc'], xlabels=[str(i) for i in np.arange(1,11)],
19 ylabels=labels, title='Coefficient matrix')
20
21def plot_matrix(ax, mat, xlabels=None, ylabels=None, title = None, xlabelrot=90):
22 """
23 Produce plot of matrix
24 """
25 ax.imshow(mat, vmin=-1, vmax=1, cmap='PiYG')
26 for i in range(mat.shape[0]):
27 for j in range(mat.shape[1]):
28 ax.text(i, j, '%.2f' % mat[j,i], ha='center', va='center',
29 fontsize=7)
30 ax.set_xticks([])
31 ax.set_yticks([])
32 ax.hlines(np.arange(1, mat.shape[0])-0.5, -0.5, mat.shape[0] - 0.5,
33 color='black', linestyle='--', linewidth=1)
34 ax.vlines(np.arange(1, mat.shape[0])-0.5, -0.5, mat.shape[0] - 0.5,
35 color='black', linestyle='--', linewidth=1)
36
37 ax.set_xticks(np.arange(0, mat.shape[0]))
38 if xlabels is not None:
39 ax.set_xticklabels(xlabels, rotation=xlabelrot)
40 ax.set_yticks(np.arange(0, mat.shape[0]))
41
42 if ylabels is not None:
43 ax.set_yticklabels(ylabels, rotation=0)
44 ax.tick_params(axis='both', which='major', labelsize=7)
45
46 if title is not None:
47 ax.set_title(title)
48
49if __name__ == '__main__':
50 main()
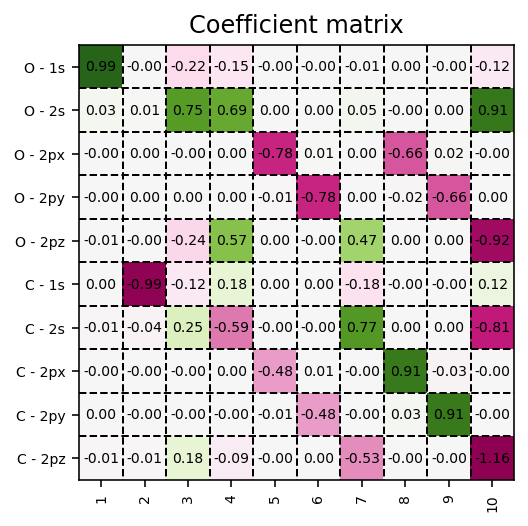
Molecular-orbital coefficient matrix in the atomic-orbital basis.
Contour plots
These orbitals can be readily visualized, as exemplified in the code below. Here,
we make use of the pydft.MolecularGrid.get_amplitude_at_points() method.
To use this method, we first have to retrieve the pydft.MolecularGrid
object from the pydft.DFT class via its pydft.DFT.get_molgrid_copy()
method.
1import numpy as np
2from pydft import MoleculeBuilder, DFT
3import matplotlib.pyplot as plt
4from mpl_toolkits.axes_grid1 import make_axes_locatable
5
6def main():
7 # perform DFT calculation on the CO molecule
8 co = MoleculeBuilder().from_name("CO")
9 dft = DFT(co, basis='sto3g')
10 res = dft.scf(1e-6, verbose=False)
11
12 # grab molecular orbital energies and coefficients
13 orbc = res['orbc']
14 orbe = res['orbe']
15
16 # generate grid of points and calculate the electron density for these points
17 sz = 4 # size of the domain
18 npts = 100 # number of sampling points per cartesian direction
19
20 # produce meshgrid for the xz-plane
21 x = np.linspace(-sz/2,sz/2,npts)
22 zz, xx = np.meshgrid(x, x, indexing='ij')
23 gridpoints = np.zeros((zz.shape[0], xx.shape[1], 3))
24 gridpoints[:,:,0] = xx
25 gridpoints[:,:,2] = zz
26 gridpoints[:,:,1] = np.zeros_like(xx) # set y-values to 0
27 gridpoints = gridpoints.reshape((-1,3))
28
29 # grab a copy of the MolecularGrid object
30 molgrid = dft.get_molgrid_copy()
31
32 fig, ax = plt.subplots(2, 5, dpi=144, figsize=(16,6))
33 for i in range(len(orbe)):
34 m_ax = ax[i//5, i%5]
35
36 # calculate field
37 field = molgrid.get_amplitude_at_points(gridpoints, orbc[:,i]).reshape((npts, npts))
38
39 # plot field
40 levels = np.linspace(-2,2,17, endpoint=True)
41 im = m_ax.contourf(x, x, field, levels=levels, cmap='PiYG')
42 m_ax.contour(x, x, field, colors='black')
43 m_ax.set_aspect('equal', 'box')
44 m_ax.set_xlabel('x-coordinate [a.u.]')
45 m_ax.set_ylabel('z-coordinate [a.u.]')
46 divider = make_axes_locatable(m_ax)
47 cax = divider.append_axes('right', size='5%', pad=0.05)
48 fig.colorbar(im, cax=cax, orientation='vertical')
49 m_ax.set_title('MO %i: %6.4f Ht' % (i+1, orbe[i]))
50
51 plt.tight_layout()
52
53if __name__ == '__main__':
54 main()
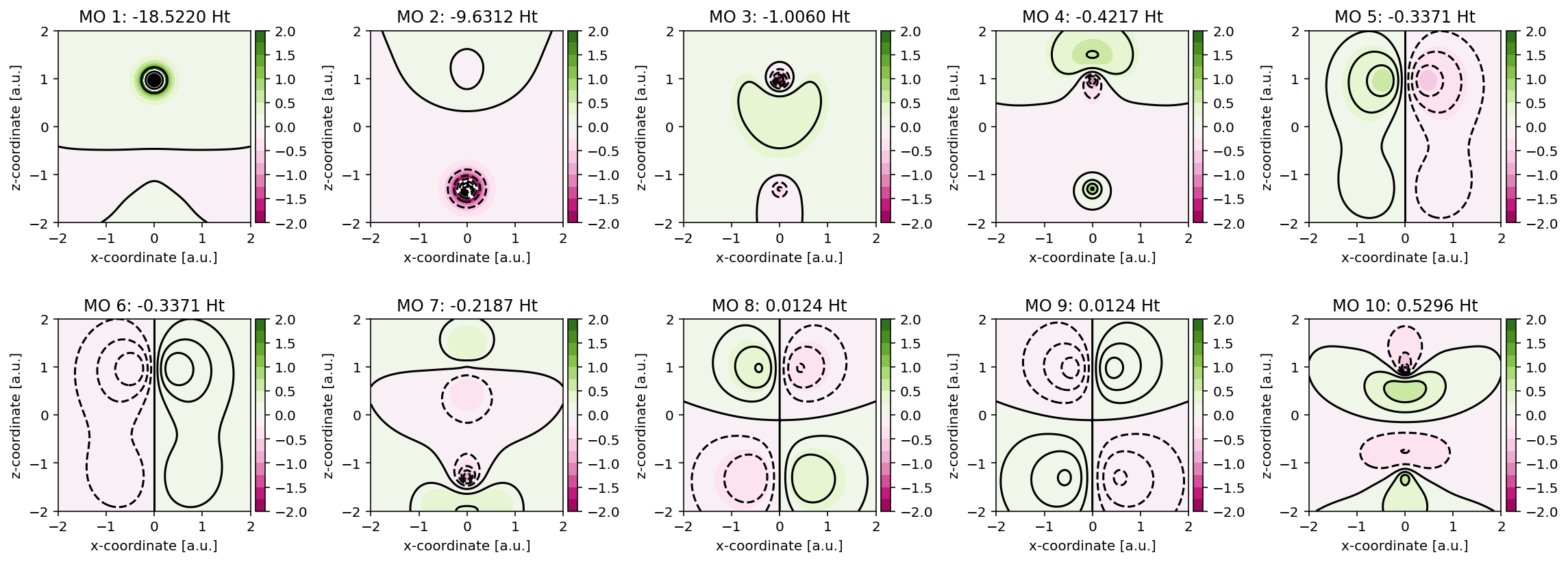
Contour plots for the Kohn-Sham molecular orbitals of CO.
See also
To get a better impression of the three-dimensional shape of the molecular orbitals, it is recommended to produce isosurfaces rather than contour plots.
Isosurfaces
Generating an isosurface is very similar to generating a contour plot, but with the notable difference that the orbital has to be sampled in three-dimensional space. In the script below, an example is provided for one of the \(1\pi\) orbitals of CO. Observe that for generating an isosurface, the algorithm has to be executed twice, once for the positive lobe and once for the negative lobe. The isosurfaces are stored in so-called Polygon File Format files which can be used in your favorite rendering program.
Note
Isosurface generation requires the PyTessel package to be installed. More information can be found here.
1import numpy as np
2from pydft import MoleculeBuilder, DFT
3from pytessel import PyTessel
4
5def main():
6 # perform DFT calculation on the CO molecule
7 co = MoleculeBuilder().from_name("CO")
8 dft = DFT(co, basis='sto3g')
9 res = dft.scf(1e-4, verbose=False)
10
11 # grab a copy of the MolecularGrid object and of the coefficient matrix
12 molgrid = dft.get_molgrid_copy()
13 orbc = res['orbc']
14
15 sz = 10 # size of the domain
16 npts = 50 # number of sampling points per cartesian direction
17
18 build_isosurface('1pi_pos.ply', molgrid, orbc[:,4], 0.03, sz, npts)
19 build_isosurface('1pi_neg.ply', molgrid, orbc[:,4], -0.03, sz, npts)
20
21def build_isosurface(filename, molgrid, coeff, isovalue, sz, npts):
22 # produce meshgrid for the unit cell
23 x = np.linspace(-sz/2, sz/2, npts)
24 zz, yy, xx = np.meshgrid(x, x, x, indexing='ij')
25 N = len(x)
26 gridpoints = np.zeros((N, N, N, 3))
27 gridpoints[:,:,:,0] = xx
28 gridpoints[:,:,:,1] = yy
29 gridpoints[:,:,:,2] = zz
30 gridpoints = gridpoints.reshape((-1,3))
31
32 # build isosurface
33 scalarfield = molgrid.get_amplitude_at_points(gridpoints, coeff).reshape(npts, npts, npts)
34 unitcell = np.diag(np.ones(3) * sz)
35
36 pytessel = PyTessel()
37 vertices, normals, indices = pytessel.marching_cubes(scalarfield.flatten(), scalarfield.shape, unitcell.flatten(), isovalue)
38 pytessel.write_ply(filename, vertices, normals, indices)
39
40if __name__ == '__main__':
41 main()