Tutorials
Converting OUTCAR to YAML file
This tutorial explains how one can parse a single VASP frequency calculation and
constructs a single .yaml-based data entry from this calculation.
Prerequisites
First, we need to prepare an OUTCAR file that we wish to parse. Several
example OUTCAR are stored in tests/data which we can unpack. For
this tutorial, we assume that we are working under examples/individual.
Go to this folder
cd examples/individual
and run
unzip ../../tests/data/OUTCAR_Ru1121_CH.zip
This should give the following output:
Archive: ../../tests/data/OUTCAR_Ru1121_CH.zip
inflating: OUTCAR_Ru1121_CH
Building YAML files
The OUTCAR_Ru1121_CH file contains a frequency calculation of a CH
molecule adsorbed on a Ru(11-21) surface. To build the yaml file, we run
pymkmkit freq2yaml OUTCAR_Ru1121_CH -o ru1121_c.yaml --average-pairs
The option --average-pairs is needed in this case because the
OUTCAR file contains a metallic slab on which CH has been adsorbed on
both sides. Thus, when sampling the frequencies for a single CH adsorbate, we
need to average out the frequencies of both the top and bottom CH fragment.
Note
The --average-pairs option is required for some older slab calculations.
Historically, adsorbates were placed on both sides of a metal slab to
cancel dipole moments before dipole corrections became standard in VASP.
Modern calculations typically use dipole corrections instead and only place
the adsorbate on one side.
In these legacy setups, two identical adsorbates (top and bottom) produce duplicate vibrational modes. Because frequencies are computed using a finite-difference approach, small numerical differences appear between the paired modes. Averaging the pairs recovers the vibrational frequencies corresponding to a single adsorbate while reducing numerical noise.
Tip
To visualize the frequency calculation and inspect vibrational modes, you can use Atom Architect. Visualizing the displacements can help identify paired modes and confirm the averaging.
Understanding the generated YAML file
The generated YAML file stores all information required to reproduce and reuse the thermodynamic state in a microkinetic model.
Below we highlight the key sections and why they are stored.
pymkmkit
Metadata describing how the file was created.
pymkmkit:
version: 0.1.0
generated: 2026-02-26T07:21:02Z
Field |
Purpose |
|
pymkmkit version used to generate the file |
|
Timestamp for provenance and reproducibility |
This metadata ensures traceability and helps diagnose differences between datasets.
structure
Describes the atomic structure used in the calculation.
structure:
formula: Ru48C2H2
lattice_vectors: [...]
coordinates_direct: [...]
pbc: [true, true, true]
Field |
Purpose |
|
Quick identification of system composition and size |
|
Defines the periodic simulation cell |
|
Atomic positions in fractional coordinates |
|
Periodic boundary conditions |
Storing the full structure allows the system to be reconstructed and verified.
calculation
Records how the calculation was performed.
calculation:
code: VASP
type: frequency
incar: {...}
potcar:
- PAW_PBE Ru
- PAW_PBE C
- PAW_PBE H
Field |
Purpose |
|
Simulation software and calculation type |
|
Important settings affecting accuracy and results |
|
Pseudopotentials used |
These parameters are essential for reproducibility and validation.
energy
energy:
electronic: -433.134185
The electronic energy forms the basis for adsorption energies, reaction energies, and activation barriers.
vibrations
Contains vibrational data required to compute thermodynamic properties.
vibrations:
frequencies_cm-1:
- 2886.2
- 714.1
partial_hessian:
dof_labels:
- 49X
- 49Y
- 49Z
- 50X
- 50Y
- 50Z
- 51X
- 51Y
- 51Z
- 52X
- 52Y
- 52Z
matrix: [...]
paired_modes_averaged: true
Field |
Purpose |
|
Vibrational modes used in partition functions |
|
Hessian submatrix for adsorbate degrees of freedom (stored for reuse) |
|
Maps each Hessian row/column to an atom index and Cartesian direction (e.g. |
|
Indicates symmetric slab modes were averaged |
Vibrational data enable computation of:
zero-point energies
entropy and heat capacity
pre-exponential factors in rate constants
The dof_labels make the partial Hessian interpretable: they encode exactly
which atoms were perturbed and in which Cartesian direction when constructing
the finite-difference Hessian, so the stored matrix can be validated,
visualized, or reprocessed later.
Tip
YAML files generated by pymkmkit can be visualized using Atom Architect. The program can display structures, vibrational modes, and displaced coordinates, making it easier to inspect the finite-difference perturbations and verify the results.
Building network.yaml file
This section explains how to build a reaction network file, using
examples/Ru1121/network.yaml as an example.
The file has four key sections:
stable_statestransition_statesnetwork(elementary reaction steps)paths(named combinations of elementary reaction steps)
Stable and transition states
The first two set of entries in the yaml file correspond to the
stable_states and transition_states. The names are
self-explanatory. Both sections are lists of YAML references. Each entry
defines:
name: shorthand (unique) label used later in step definitionsfile: relative path to the corresponding state YAML file
Examples
For example, in examples/Ru1121/network.yaml we find:
stable_states:
- name: CO*
file: ISFS/co.yaml
- name: C*
file: ISFS/c.yaml
transition_states:
- name: CO_diss
file: TS/co_diss.yaml
This defines two stable states, corresponding to CO* and C* adsorbed on the
catalytic surface whose state is defined by the YAML files
ISFS/co.yaml and TS/co_diss.yaml, respectively.
Defining elementary reaction steps
The network section is a list of elementary steps. Each step has:
name: unique step identifiertype:surforadsreaction: human-readable reaction string
For surf steps, energies are given through forward and backward
blocks, each containing:
ts: list of transition-state termsis: list of initial-state termsnormalization: divisor used to normalize to one adsorbate/site unit
Each ts or is term has:
name: label fromstable_statesortransition_statesstoichiometry: multiplicative factor
For ads steps, the file uses is and fs (initial/final state) plus
normalization.
The normalization entry is used when calculations contain multiple
symmetry-equivalent adsorbates (e.g., adsorbates on both slab faces). Since the
computed energies and ZPE contributions correspond to the combined system,
normalization: 2 converts them to per-adsorbate values.
How pymkmkit evaluates barriers (including ZPE)
For a surface step direction (forward or backward), pymkmkit computes:
with
implemented from vibrational frequencies in cm-1 as
Examples
CO dissociation
- name: CO dissociation
type: surf
reaction: CO* + * => C* + O*
forward:
ts:
- name: CO_diss
stoichiometry: 1
is:
- name: CO*
stoichiometry: 1
normalization: 2
Forward barrier equations are:
C hydrogenation
- name: C hydrogenation
type: surf
reaction: C* + H* => CH* + *
forward:
ts:
- name: C_hydr
stoichiometry: 1
- name: empty
stoichiometry: 1
is:
- name: C*
stoichiometry: 1
- name: H*
stoichiometry: 1
normalization: 2
Forward barrier equations are:
Grouping elementary steps into mechanisms using paths
The paths section defines named sequences of network steps, for example to
evaluate overall reaction energies or to build potential energy diagrams.
Each path contains:
name: path identifierstartlabel: label shown for the initial state in PED outputsteps: ordered list of elementary steps byname
Optional per-step entries:
factor(default1): multiply that step contribution; this is used to represent successive applications of this elementary reaction step, for example when you want sixfold 1/2 H2 adsorption.label: custom state label for PED annotations
Example (from the Ru(11-21) case):
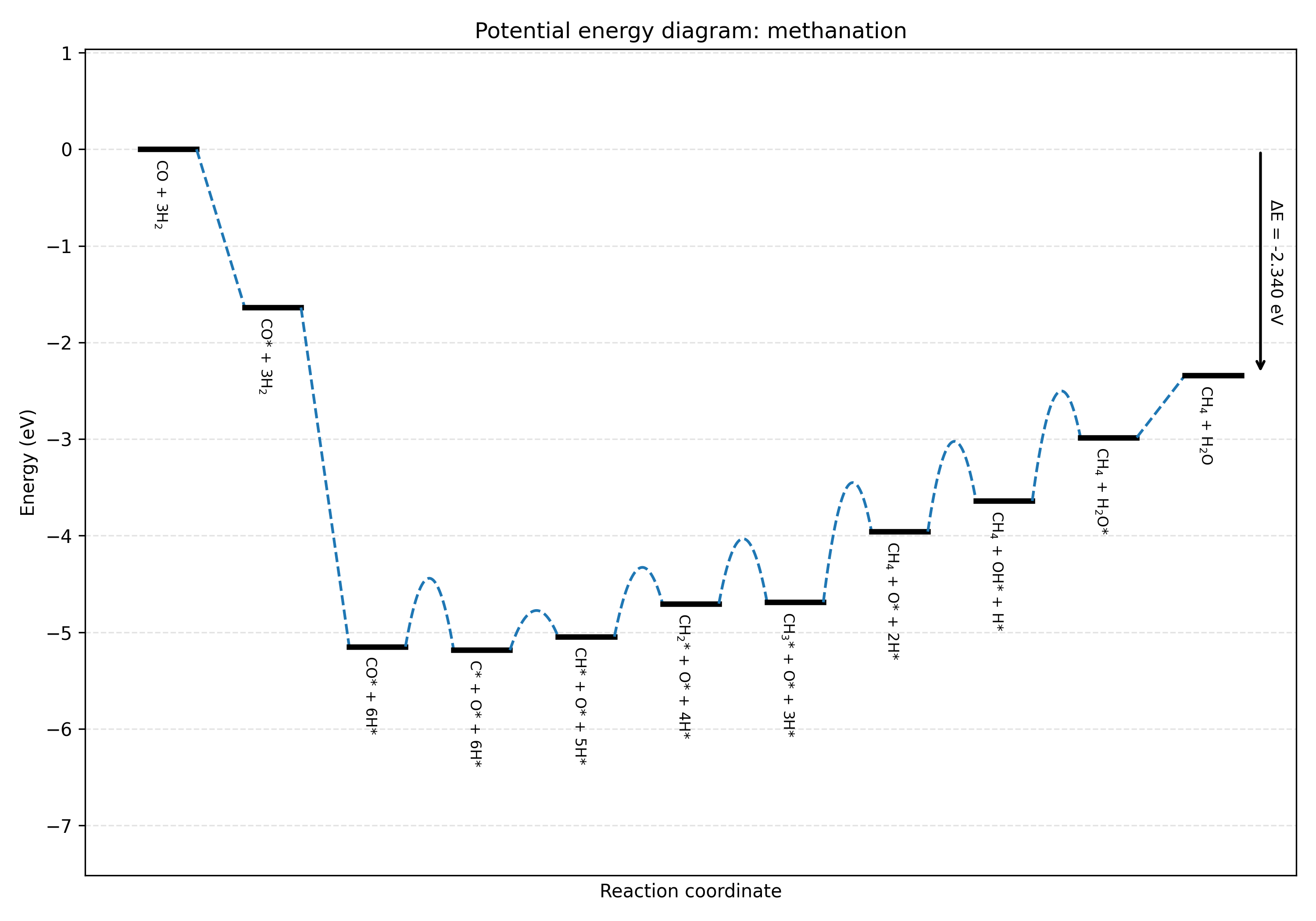
paths:
- name: methanation
startlabel: CO + 3H2
steps:
- name: CO adsorption
label: CO* + 3H2
- name: H2 adsorption
factor: 6
label: CO* + 6H*
Evaluating network files
From the repository root, you can run:
pymkmkit read_network examples/Ru1121/network.yaml
This parses all states and prints step-wise quantities:
for
surfsteps: forward and reverse barriers (including ZPE correction)for
adssteps: adsorption heat (including ZPE correction)
which yields the following output:
Reaction: CO* + * => C* + O*
Forward barrier: 0.708003 eV (inc. ZPE-corr: -0.031787)
Reverse barrier: 0.743850 eV (inc. ZPE-corr: -0.024853)
Reaction: C* + H* => CH* + *
Forward barrier: 0.406754 eV (inc. ZPE-corr: -0.047930)
Reverse barrier: 0.267382 eV (inc. ZPE-corr: -0.139948)
Reaction: CH* + H* => CH2* + *
Forward barrier: 0.703780 eV (inc. ZPE-corr: -0.038874)
Reverse barrier: 0.362366 eV (inc. ZPE-corr: -0.108891)
Reaction: CH2* + H* => CH3* + *
Forward barrier: 0.672479 eV (inc. ZPE-corr: 0.046713)
Reverse barrier: 0.657561 eV (inc. ZPE-corr: -0.122669)
Reaction: CH3* + H* => CH4 + 2*
Forward barrier: 1.198466 eV (inc. ZPE-corr: -0.012794)
Reverse barrier: 0.466999 eV (inc. ZPE-corr: -0.138733)
Reaction: O* + H* => OH* + *
Forward barrier: 0.925349 eV (inc. ZPE-corr: -0.075468)
Reverse barrier: 0.607454 eV (inc. ZPE-corr: -0.197065)
Reaction: OH* + H* => H2O* + *
Forward barrier: 1.102802 eV (inc. ZPE-corr: 0.048977)
Reverse barrier: 0.446876 eV (inc. ZPE-corr: -0.068496)
Reaction: CO + * => CO*
Adsorption heat: -1.638042 eV (inc. ZPE-corr: 0.021169)
Reaction: H2 + 2* => 2H*
Adsorption heat: -0.585109 eV (inc. ZPE-corr: 0.030218)
Reaction: H2O + * => H2O*
Adsorption heat: -0.643202 eV (inc. ZPE-corr: 0.059390)
To evaluate total energies of all named paths:
pymkmkit evaluate_paths examples/Ru1121/network.yaml
This sums the (factor-weighted) step reaction heats for each path as given by the following output:
Path methanation: -2.340352 eV
To construct a potential energy diagram for one path:
pymkmkit build_ped examples/Ru1121/network.yaml methanation methanation.png
This builds the PED of the methanation path and writes it to
methanation.png.